Different projects and products from the GA4GH ecosystem are assumed to develop or adopt domain specific data models.
The original GA4GH data model - developed by GA4GH Data Working Group (DWG) - used a general object model which combined elements of the VCF structure (variants and callsets) with a commonly used representation of “biological objects” (individual, biosample) for provenance tracking and representation of “meta information” related to the individual genotyping results.
This hierarchical object model is well suited for the representation of data from individuals and their genotyping information. It had not been developed to document e.g. recurring evidence documentation or equivalence modeling of their physiologic and phenotypic associations.
This general object model is used in various implementations, with some
variations regarding requirements for the individual components (e.g.
Phenopackets may not make use of the biosample component in a germline/rare
disease setting; Beacon resources may not link up to individuals or even
biosamples in their aggregate backend version).
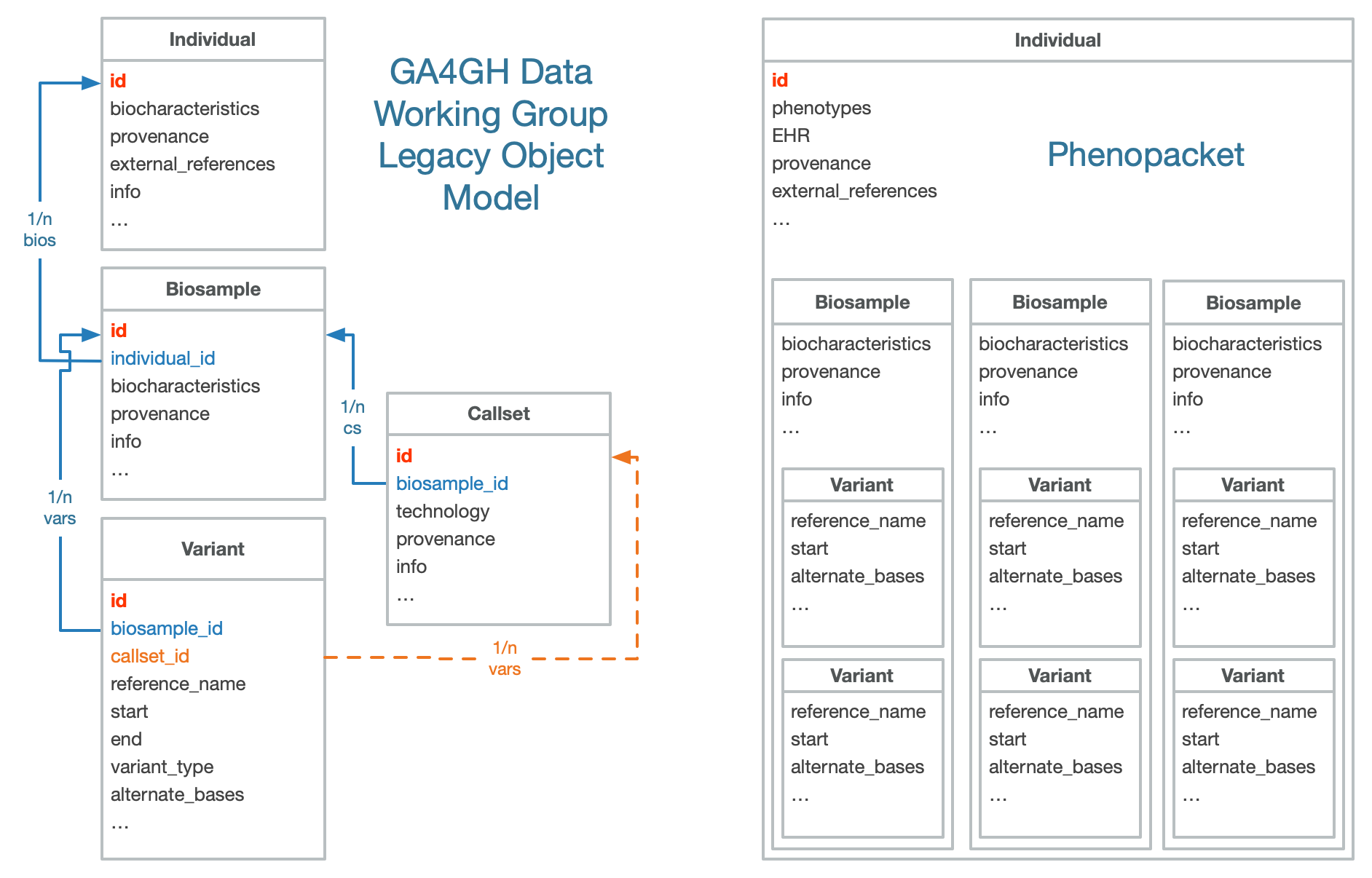
The GA4GH data model for genomics recommends the use of a principle object hierarchy, consisting of
variant
biosamplecallset (also analysis or several technical objects)
biosamplecallset can be compared to a data column in a VCF variant annotation filecallset has an optional position in the object hierarchy, since variants describe biological observations in a biosample and can be seen as the entity describing the technologies and analysis procedures leading from the sample to the set of all variantsbiosample
individual (also subject)
Additional concepts (e.g. dataset, study …) may be added in the future.
In the design of genomics APIs, file formats and storage protocols, it is of relevance to adhere to a logical object structure which reflects physical reality and common data handling procedures.
At the core of many (human health related and other) databases and procedural systems is the concept of a “biosample”, representing the source of biological material on which some (molecular or other) analyses are being performed, leading to a set of observations (e.g. the genomic variants measured by Whole Genome Sequencing and called against a reference genome, in the DNA extracted from a tissue biopsy).
For a consistant API design, it is important to relate observations and measurement to the correct object in the data model’s hierarchy. A typical example human genomic data analysis is the association of phenotypic information to the type of biosample being analysed. For the association of genomic variants with a cancer diagnosis, it is of paramount importance to know if - for an individual with a cancer diagnosis - the observed variants were called from a germline biosample (i.e. analysis of cancer predisposition) or from a cancer tissue biosample (i.e. somatic mutation analysis).
